BioMASS
![]()




![]()
Mathematical modeling is a powerful method for the analysis of complex biological systems. Although there are many researches devoted on producing models to describe dynamical cellular signaling systems, most of these models are limited and do not cover multiple pathways. Therefore, there is a challenge to combine these models to enable understanding at a larger scale. Nevertheless, larger network means that it gets more difficult to estimate parameters to reproduce dynamic experimental data needed for deeper understanding of a system.
To overcome this problem, we developed BioMASS, a Python framework for Modeling and Analysis of Signaling Systems. The BioMASS framework allows efficient optimization of multiple parameter sets simultaneously and generates the multiple parameter candidates that explain the signaling dynamics of interest. These parameter candidates can be further evaluated by their distribution and sensitivity analysis as a part of alternative information about the hidden regulatory mechanism of the system.
Features
- Parameter estimation of ODE models
- Local sensitivity analysis
- Effective visualization of simulation results
Documentation
Online documentation is available at https://biomass-core.readthedocs.io/.
Installation
The BioMASS library is available at the Python Package Index (PyPI).
$ pip install biomass
BioMASS supports Python 3.7 or newer.
Also, we provide BioMASS docker images on DockerHub.
Example
Parameter estimation
from biomass import Model, optimize
from biomass.models import Nakakuki_Cell_2010
model = Model(Nakakuki_Cell_2010.__package__).create()
optimize(model, x_id=range(1, 11))
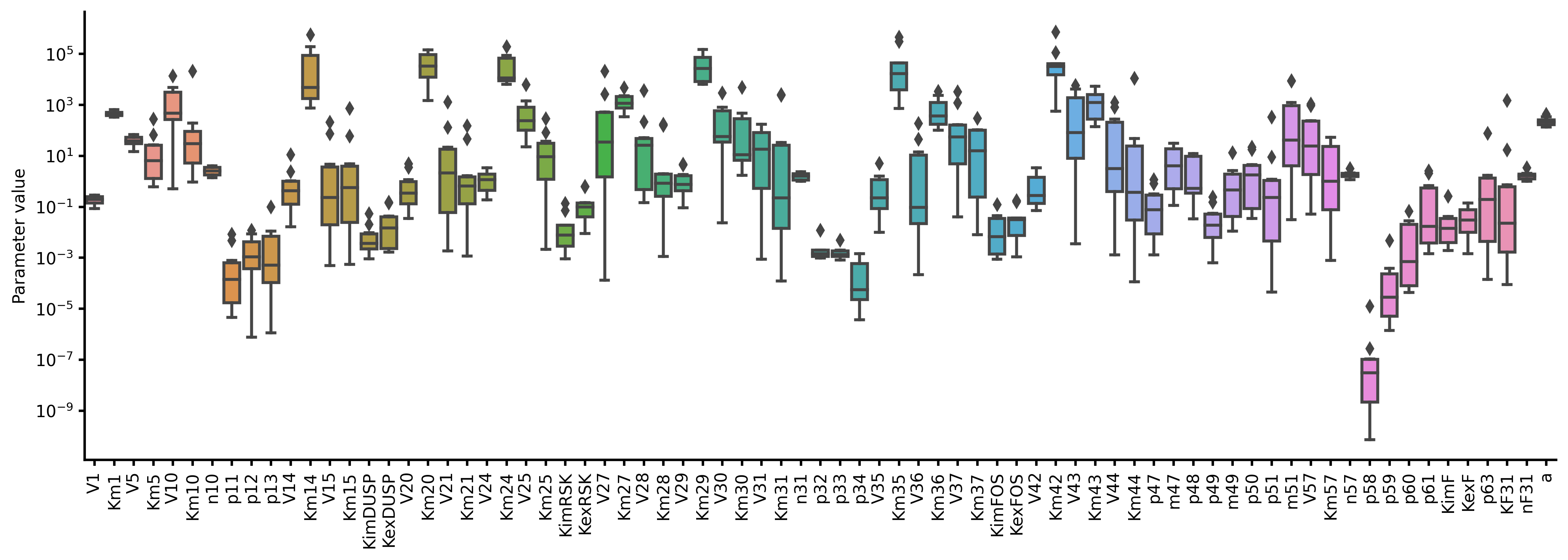
from biomass import run_simulation
run_simulation(model, viz_type="average", stdev=True)
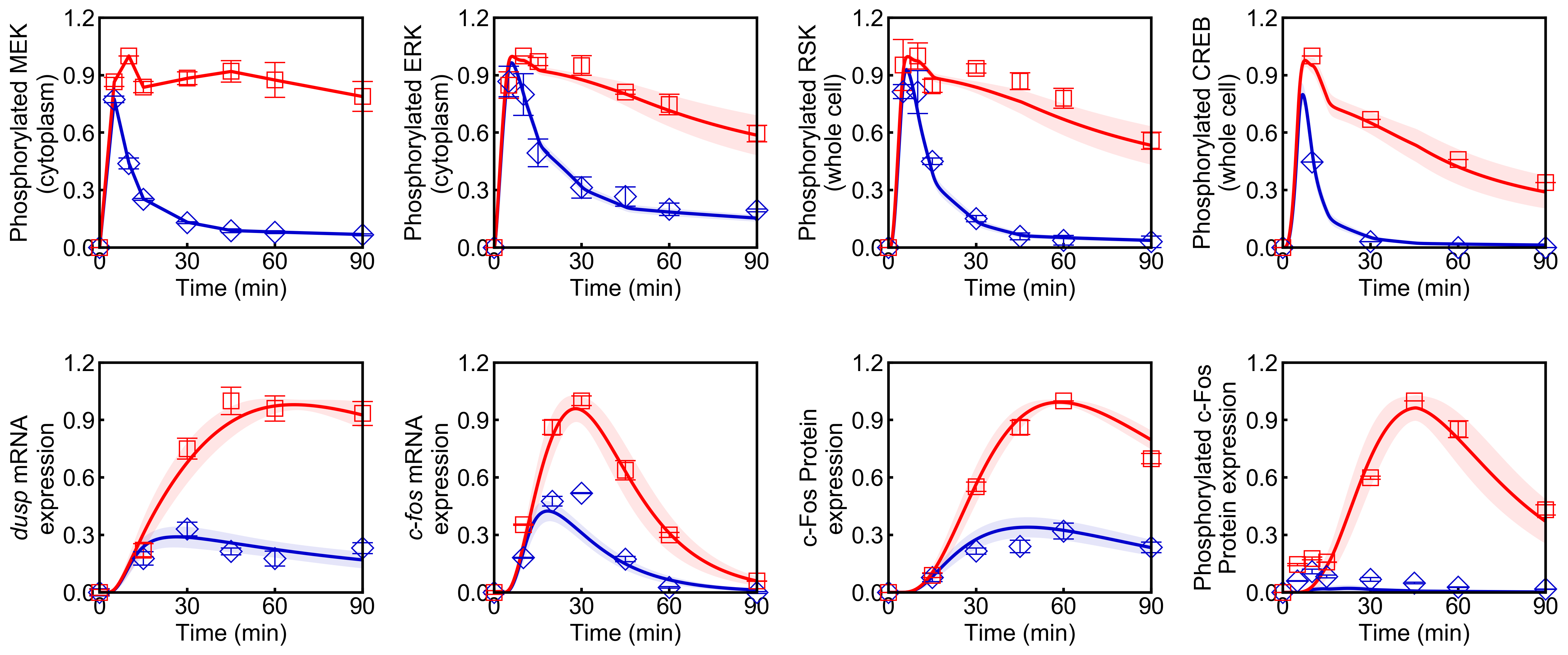 Points (blue diamonds, EGF; red squares, HRG) denote experimental data, solid lines denote simulations.
Points (blue diamonds, EGF; red squares, HRG) denote experimental data, solid lines denote simulations.
Sensitivity analysis
from biomass import run_analysis
run_analysis(model, target="reaction", metric="integral", style="barplot")
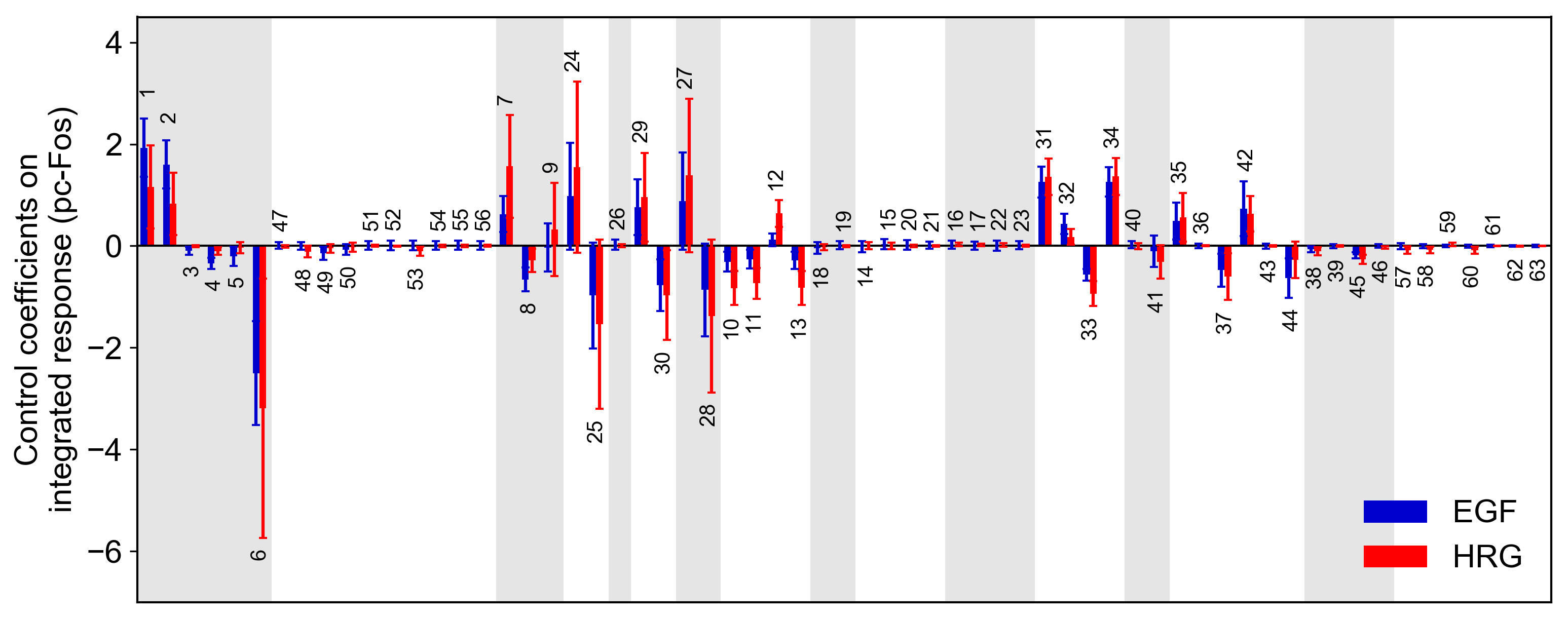
Control coefficients for integrated pc-Fos are shown by bars (blue, EGF; red, HRG). Numbers above bars indicate the reaction indices, and error bars correspond to simulation standard deviation.
Citation
When using BioMASS, please cite the following paper:
- Imoto, H., Zhang, S. & Okada, M. A Computational Framework for Prediction and Analysis of Cancer Signaling Dynamics from RNA Sequencing Data—Application to the ErbB Receptor Signaling Pathway. Cancers 12, 2878 (2020). https://doi.org/10.3390/cancers12102878






 114 Dec 23, 2022
114 Dec 23, 2022
 23 Dec 16, 2022
23 Dec 16, 2022
 1 Dec 08, 2021
1 Dec 08, 2021
 4 Mar 06, 2022
4 Mar 06, 2022
 2 Nov 16, 2021
2 Nov 16, 2021
 1 Jan 26, 2022
1 Jan 26, 2022
 1 Dec 09, 2021
1 Dec 09, 2021
 179 Dec 21, 2022
179 Dec 21, 2022
 2.8k Dec 31, 2022
2.8k Dec 31, 2022
 1 Jan 12, 2022
1 Jan 12, 2022
 1 Oct 02, 2022
1 Oct 02, 2022
 27 Dec 09, 2022
27 Dec 09, 2022
 73 Jan 03, 2023
73 Jan 03, 2023
 3 Nov 16, 2021
3 Nov 16, 2021
 3.7k Jan 03, 2023
3.7k Jan 03, 2023
 136 Dec 22, 2022
136 Dec 22, 2022
 762 Jan 07, 2023
762 Jan 07, 2023
 2 Jan 20, 2022
2 Jan 20, 2022
 76 Nov 30, 2022
76 Nov 30, 2022
 11 Dec 30, 2022
11 Dec 30, 2022